breakaway is a package for species richness estimation
and modelling. As the package has grown and users have requested more
functionality, it contains some basic generalisations of the statistical
philosophy that underpins breakaway to the general alpha
diversity case. Because of the flexibility of the modelling strategies,
most users of breakaway are microbial ecologists with very large OTU
tables, however, nothing should exclude a macroecologist from using the
same tools. If you have a macroecology dataset and want to use this
package, I would love to hear from you so please feel free to contact me
(email or via Github’s Issues/Projects infrastructure).
Vignette Info
This vignette will lead you through the most basic way to use
breakaway. For a more in depth discussion of how and why
estimating species richness is possible, check out the Introduction
to diversity estimation vignette.
Creating frequency tables
We’re now going to “collapse” the otu_data’s columns (samples) into frequency tables. Frequency tables…
frequencytablelist <- build_frequency_count_tables(otu_data)
head(frequencytablelist[[63]])## index frequency
## 1 1 35
## 2 2 22
## 3 3 15
## 4 4 17
## 5 5 11
## 6 6 10Interpretation: In this sample, there were 35 different species observed only once (singletons), 22 different species observed only twice, …, 1 species observed 2922 times.
Estimating species richness
Let’s run breakaway on the first frequency count table
breakaway(frequencytablelist[[1]])## Estimate of richness from method breakaway:
## Estimate is 359
## with standard error 325.81
## Confidence interval: (262, 2402)You should get some output to screen, including your estimate & s.e. You can also investigate a plot of the fits to the ratios as follows:
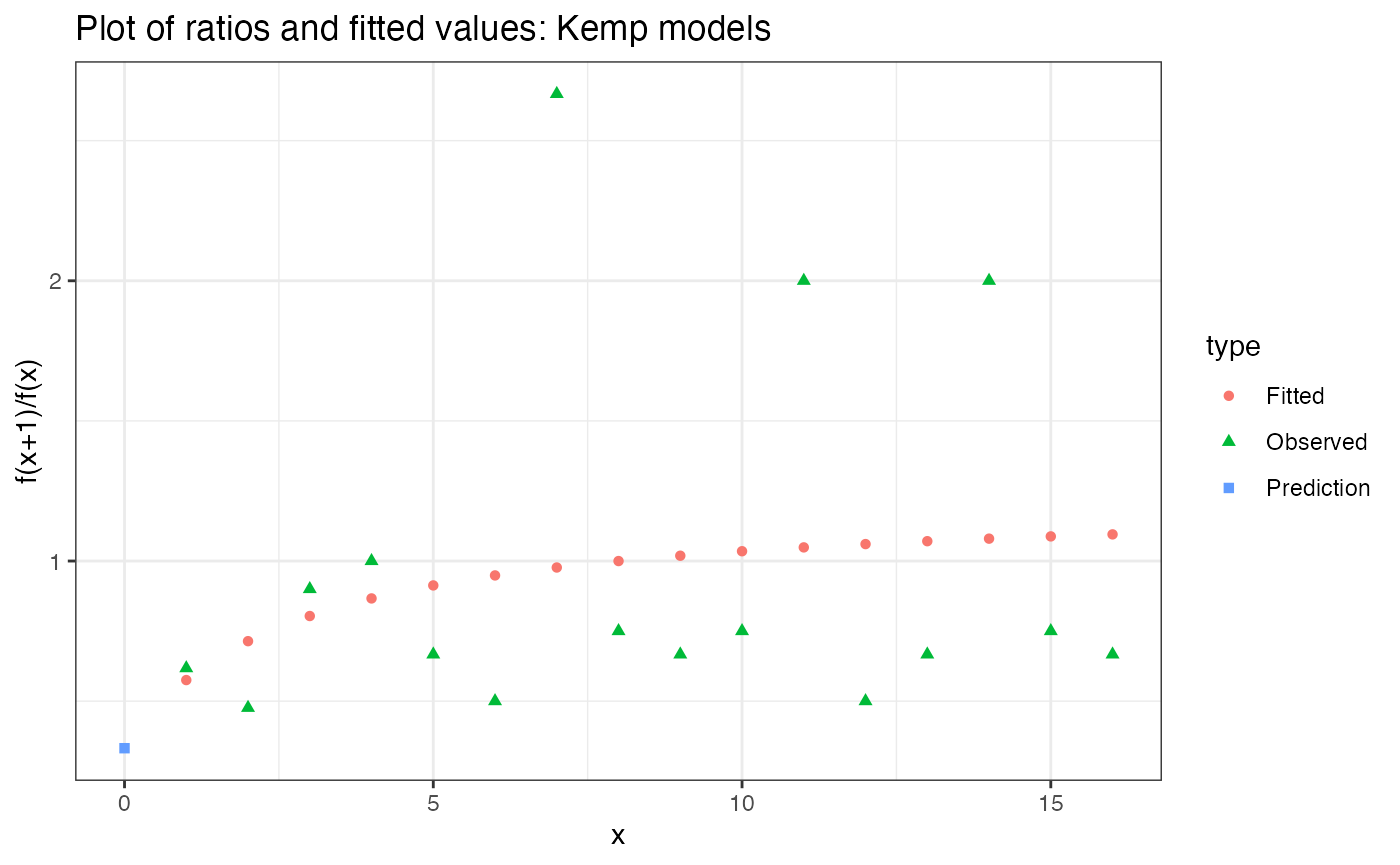
Note that it is not a fit to the frequencies, it is a fit to the ratios of frequencies. You would never need to include this type of plot in one of your papers. It is solely for you to check for model misspecification. What’s model misspecification? If the model fit (pink circles) don’t remotely follow the pattern of the observed ratios (green triangles), that’s model misspecification.
Sometimes, breakaway’s usual procedure doesn’t work, that is, it
gives a negative estimate, which is of course silly. In that case,
breakaway returns a different model’s result. It’s called the WLRM.
There isn’t a picture. Only some of the models will provide plots, and
if the model chosen does not include a plot, the plot function will
instead return NULL. Here is an example of a case where
breakaway returns the WLRM.
breakaway(frequencytablelist[[2]])## Estimate of richness from method breakaway:
## Estimate is 346
## with standard error 42.82
## Confidence interval: (301, 492)
## Cutoff: 15breakaway can defer to the WLRM for several reasons. Perhaps there
are too many singletons. Perhaps there isn’t a long enough tail. Perhaps
there is false diversity. Regardless, we can see if this failure was
sensitive to the singleton count by running
breakaway_nof1(). This requires no singleton count
(implicit is that the singleton count was erroneous) and predicts it
from the other frequencies:
breakaway_nof1(frequencytablelist[[2]][-1,])## Estimate of richness from method PoissonModel:
## Estimate is 75
## with standard error 0.47
## Confidence interval: (75, 77)The reference for breakaway_nof1() is: Willis, A.
(2016). Species richness estimation with high diversity but spurious
singletons.
breakaway_nof1 is an exploratory tool for assessing sensitivity of breakaway to the singleton count. I recommend it as a sensitivity analysis rather than for diversity estimation.
Next steps
- The above discussion focused exclusively on richness. Check out
github.com/adw96/DivNetfor alpha and beta diversity tutorials! - To learn about modelling alpha diversity, see the vignette Introduction to hypothesis testing for diversity
- To hear more details about how species richness estimation works, check out Introduction to diversity estimation